The clot thickens: Thromboembolic disease in dogs and cats
Nicole Barrella, DVM
Massachusetts Veterinary Referral Hospital, Woburn, MA
Posted on 2016-10-18
Hemostasis
The cell-based model of coagulation describes the three distinct, overlapping phases leading to clot formation as a result of platelet interaction with tissue factor (TF). The first phase, initiation, occurs when a TF-bearing cell, such as a sub-endothelial cell, is exposed to flowing blood. Circulating factor VIIa (FVIIa) binds to TF and initiates the coagulation cascade resulting in thrombin formation. The thrombin binds to receptors on the surface of the platelet leading to platelet shape change, creation of a procoagulant membrane surface, and platelet granule release. The granules contain agonists, such as thromboxane, von Willebrand’s factor (vWF) and ADP to amplify platelet activation and cause aggregation of platelets. Fibrinogen is also released and binds to integrin receptor αIIbβ3 which mediates platelet-platelet interactions. This is the amplification phase. The final phase is propagation, which results in the recruitment of additional platelets to the site of injury, and continued thrombin formation via cleavage of prothrombin to thrombin by factors Va and Xa. As the concentration of thrombin increases, there is increased cleavage of fibrin from fibrinogen. The soluble fibrin molecules will polymerize into fibrin strands and become an insoluble fibrin matrix.
Fibrinolysis begins when clot formation is complete to limit the extent of the clot and to breakdown the clot. In the presence of fibrin, plasminogen is converted to plasmin by tissue plasminogen activator (t-PA). Plasmin degrades fibrinogen and soluble fibrin into fibrinogen/fibrin degradation products (FDPs) and degrades cross-linked fibrin into D-dimers.
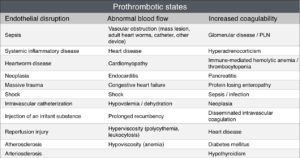
Virchow’s triad describes three factors involved in thrombus formation – blood stasis, endothelial disturbances, and hypercoagulability – with an alteration in any of the three resulting in a blood clot.
Endothelial Disturbances
Endothelial dysfunction leads to numerous changes that ultimately alter the coagulability of blood. Damage to the endothelium disrupts the anti-coagulant, anti-platelet and fibrinolytic effects that serve to prevent pathological clot formation. The endothelium consists of vascular endothelial cells and the glycocalyx layer. The glycocalyx contains anticoagulant elements and acts as a mechanoreceptor leading to release of nitric oxide in conditions of increased shear stress. The glycocalyx also acts as a shield preventing binding of inflammatory mediators onto the cell surface.
An endothelial disturbance can occur secondary to injury from high shear conditions, such as in cases of hypertension or turbulent blood flow, or due to activation by inflammatory mediators such as tumor necrosis factor-α, bradykinin, and histamine.
Upon activation or injury, endothelial cells release ultra large multimers of vWF, which can initiate platelet activation and adhesion. In healthy patients, these multimers are broken down into smaller components by the enzyme ADAMTS13. Lower concentrations of ADAMTS13 can lead to systemic platelet aggregation and thrombosis.
Decreased Endogenous Anticoagulant Elements
The three main endogenous anticoagulants are antithrombin (AT), protein C, and tissue factor pathway inhibitor (TFPI). Antithrombin inhibits thrombin and factor Xa, and is most effective when bound to endogenous or exogenous heparins. Antithrombin can be decreased secondary to decreased production, consumption, or degradation by neutrophils. Antithrombin is a small protein and can be lost into urine in cases of glomerulonephritis or, less frequently, into the intestines in cases of protein-losing enteropathy. Protein C inhibits factors Va and VIIIa, decreasing the breakdown of prothrombin to thrombin. Activated protein C also enhances fibrinolysis by suppressing plasminogen activator inhibitor-1 activity and thrombin activatable fibrinolytic inhibitor activity, resulting in increased conversion of plasminogen to plasmin. In patients with systemic inflammation, there is decreased Protein C production. Additionally, activation of Protein C is decreased by the effects of inflammatory cytokines on the endothelium.
Diagnosis of a hypercoagulable state
Identification of an underlying procoagulant disease process is the first step in identifying a hypercoagulable patient. Diagnosis of hypercoagulability prior to a thrombotic event can be challenging. Traditional coagulation testing, including prothrombin time (PT), activated partial thromboplastin time (aPTT), and platelet count are more accurate for hypocoaguability. Viscoelastic coagulation testing, such as thromboelastography (TEG), is considered a more global assessment of coagulation. This diagnostic test assesses duration until clot formation begins, the speed of clot formation, clot stability, and speed of fibrinolysis. Hypercoagulable samples have a faster rate of clot formation and greater clot strength than normocoagulable samples. Fibrinolysis may also be inhibited.
Platelet function is assessed in whole blood viscoelastic testing. Markers of platelet activation, such as P-selectin expression or increased platelet reactivity can also be measured. Aggregation assays and platelet function analyzers are more often used in diagnosis of primary hemostatic disturbances. Platelet agonists are used to initiate clot formation. Shortened closure time, as determined by a platelet function analyzer, has not been linked to thrombosis in veterinary patients. The mean platelet component estimates the refractive index of platelets, with activated platelets having a decreased density due to release of granule contents.
Procoagulant, anti-coagulant and fibrinolytic components can be measured to document hypercoagulability. An increase in procoagulant elements (factor V or VIII activity, fibrinogen concentration), a decrease in endogenous anticoagulants (antithrombin concentration, Protein C activity), or a decrease in fibrinolytic components (t-PA concentrations, plasminogen concentrations) all support a hypercoagulable state. These tests can be performed at a specialty reference laboratory, and are useful in confirmation, however they are usually not practical in clinical situations.
Diagnosis of Thrombosis
Clinical findings of thromboembolic disease depend on the type and location of vessels obstructed, the degree of vascular obstruction, and the extent of associated tissue damage. Common locations for arterial thrombi include the distal aorta, brachial and pulmonary arteries. Arterial thromboembolism is more common in cats than in dogs. The most common cause of arterial thromboembolism in cats is cardiomyopathy, with neoplastic and systemic inflammation being less common causes. Distal aortic thromboembolism (ATE) can be diagnosed by comparing blood glucose concentrations. In a study evaluating peripheral blood glucose concentration of the affected limb and central blood glucose concentration, the affected limb had a significantly lower blood glucose concentration in cats (median difference of 107 mg/dL in the definitely diagnosed ATE group) and dogs (median difference of 46 mg/dL) when compared to ambulatory and non-ambulatory controls.
In dogs, arterial thromboembolism has been associated with PLNs, hyperadrenocorticism, neoplasia, hypothyroidism, GDV, pancreatitis and cardiovascular disease. Pulmonary thromboembolism (PTE) is suspected in patients with hypoxemia, hypocapnia, and in increased alveolar-arterial oxygen gradient. Hypoxemia is poorly responsive to oxygen. There are no specific changes seen on thoracic radiographs that can be used to diagnose a PTE. Pulmonary angiography or computed tomographic (CT) angiography can be used to diagnose PTE. A normal pulmonary angiogram rules out PTE. Either of these tests can require general anesthesia, which may be undesirable in an unstable patient. Intra-abdominal thrombi can be seen with abdominal ultrasound.
Treatment of hypercoagulable states and thrombosis
The goals of therapy are 1) stabilization with supportive treatment of the patient, 2) prevention of thrombus expansion and the formation of additional clots, and 3) reduction in size of the existing clots and restoration of perfusion. Stabilization may involve oxygen therapy, fluid therapy, and analgesia. Management should be provided based on the individual patient’s needs.
Anticoagulant therapy in veterinary patients consists primarily of various types of heparin. Coumarin derivatives can also be used, but will not be discussed here. Heparins enhance the inhibitory activity of AT by binding to AT, which then inactivates thrombin and factor Xa. Short molecules of heparin are better at catalyzing AT to inhibit factor Xa than to inhibit thrombin. Heparins can also cause the release of TFPI from the endothelial surface. Sodium heparin is an unfractionated heparin, and contains heparin molecules of varying sizes. It is widely available and inexpensive. Unfractionated heparin has the highest variability in dose-response relationship and the greatest potential for over-anticoagulation. The recommended dose for dogs is currently 150-300 units/kg subcutaneously every 6-8 hours. For critically ill animals, a loading dose of 100 units/kg followed by a constant rate intravenous infusion of 20-50 units/kg/hr is more likely to result in consistent anti-coagulation. In cats, doses range from 50-100 unit/kg to 200-300 unit/kg every 6-8 hours for prevention of aortic thromboembolism. Monitoring of aPTT should be performed regularly, with a target prolongation of 1.5-2.5 fold. Enoxaparin and dalteparin are low molecular weight heparins, and are administered subcutaneously. They tend to have longer half-lives than unfractionated heparin, so less frequent dosing is required. Enoxaparin dosing is 0.8mg/kg subcutaneously every six hours in dogs, with no significant hemorrhagic complications reported. In cats, 1mg/kg subcutaneously every 12 hours is reported as effective. Doses for dalteparin sodium are 100-150 units/kg subcutaneously every 12-24 hours in dogs, and 100 units/kg subcutaneously every 12-24 hours in cats.
Antiplatelet drugs include aspirin and clopidogrel. Aspirin inhibits cyclooxygenase, which reduces the synthesis of prostacyclin from the endothelium and thromboxane A2 from the platelets and kidney. Significant increases in closure times on a platelet function analyzer were noted in 1/3 of dogs given aspirin 1 mg/kg orally once a day for ten days. The experiment was repeated with both in vitro and in vivo assessment of platelet function, and response among sixteen dogs varied. In cats, doses of 5mg/cat every third day has lower gastrointestinal side effects than previously reported 10mg/kg every 48-72 hours. Clopidogrel is an adenosine diphosphate (ADP) receptor antagonist. It is a prodrug that is metabolized by the cytochrome P450-dependent pathway to create active metabolites. The metabolites bind irreversibly to the ADP receptor on the platelet surface. This inhibits platelet aggregation and causes a dose-dependent reduction of platelet binding sites for ADP. Dosing starts at 1mg/kg by mouth once daily in dogs, but up to 5mg/kg can by given once daily. There is no significant evidence supporting the use of clopidogrel in dogs. In cats, the dose of 18.75mg (1/4 of 75mg tablet) by mouth once daily was supported by the FAT CAT trial.
Veterinary experience with thrombolytic therapy is limited. Drugs that are used in human medicine include streptokinase, urokinase, and human recombinant tissue plasminogen activator (rt-PA). These drugs increase conversion of plasminogen to plasmin. Complications related to their use include reperfusion injury, hemorrhage and high mortality rate. Additionally, the dosing protocols are unclear in veterinary patients, and there has not been strong evidence showing a survival advantage. Use of these agents is recommended within 3-4 hours of the event.
Abbreviations
aPTT: activated partial thromboplastin time
AT: antithrombin
ATE: aortic thromboembolism
CT: computed tomography
FDP: fibrinogen/fibrin degradation products
fVIIa: Factor VIIa (activated)
PT: prothrombin time
PTE: pulmonary thromboembolism
rt-PA: recombinant tissue plasminogen activator
t-PA: tissue plasminogen activator
TEG: thromboelastography
TF: tissue factor
TFPI: tissue factor pathway inhibitor
vWF: von Willebrand’s factor
Further reading
- De Laforcade A. Diseases associated with thrombosis. Top Companion Anim Med. 2012;27(2):59-64.
- Dudley A, Thomason J, Fritz S, et al. Cyclooxygenase Expression and Platelet Function in Healthy Dogs Receiving Low Dose Aspirin. JVIM 2013;27(1): 141-149
- Goggs R, Benigni L, Fuentes VL, Chan DL. Pulmonary thromboembolism. J Vet Emerg Crit Care (San Antonio). 2009;19(1):30-52.
- Haines JM, Thomason JM, Seage EC, Wills RW, et al. In vitro and in vivo assessment of platelet function in healthy dogs during administration of a low-dose aspirin regimen. AJVR 2016;77(2): 174-185.
- Hogan DF, Fox PR, Jacob K, et al. Secondary prevention of cardiogenic arterial thromboembolism in the cat: The double-blind, randomized, positive-controlled feline arterial thromboembolism; clopidogrel vs. aspirin trial (FAT CAT). J Vet Cardiol. 2015;17 Suppl 1:S306-17.
- Klainbart S, Kelmer E, Vidmayer B, Bdolah-abram T, Segev G, Aroch I. Peripheral and central venous blood glucose concentrations in dogs and cats with acute arterial thromboembolism. J Vet Intern Med. 2014;28(5):1513-9.
- Panek CM, Nakamura RK, Bianco D. Use of enoxaparin in dogs with primary immune-mediated hemolytic anemia: 21 cases. J Vet Emerg Crit Care (San Antonio). 2015;25(2):273-7.
- Ralph AG, Brainard BM. Hypercoagulable states. In: Silverstein D, Hopper K. Small Animal Critical Care Medicine. Saunders; 2014.
- Smith SA. Antithrombotic therapy. Top Companion Anim Med. 2012;27(2):88-94.
- Van de wiele CM, Hogan DF, Green HW, Sederquist KD. Antithrombotic effect of enoxaparin in clinically healthy cats: a venous stasis model. J Vet Intern Med. 2010;24(1):185-91.
- Ware WA. Thromboembolic disease. In: Cardiovascular Disease in Small Animal Medicine. Manson Publishing; 2011.
- Wiinberg B, Jessen LR, Tarnow I, Kristensen AT. Diagnosis and treatment of platelet hyperactivity in relation to thrombosis in dogs and cats. J Vet Emerg Crit Care 2012;22(1):42-58.
About the author
|


