Canine hypoadrenocorticism: An overview of what we know and have learned
Marion Haber, DVM, DACVIM
Massachusetts Veterinary Referral Hospital
Hypoadrenocorticism is a life-threatening disease and is a result of failure of the adrenal glands to produce adequate hormones. Eighty-five percent of both adrenal glands have to be destroyed in order for clinical signs to occur. Most commonly, deficiency of both mineralocorticoid and glucocorticoid hormones is present; however, an atypical form also occurs where deficiency of glucocorticoid hormones alone causes significant disease. In humans, Thomas Addison first described hypoadrenocorticism in 1855 where necropsies showed complete destruction of the adrenal glands in patients with clinical signs now associated with this disease. The first case of hypoadrenocorticism in dogs was described in 1953 and in cats in the 1980s.
Physiology and etiology of hypoadrenocorticism
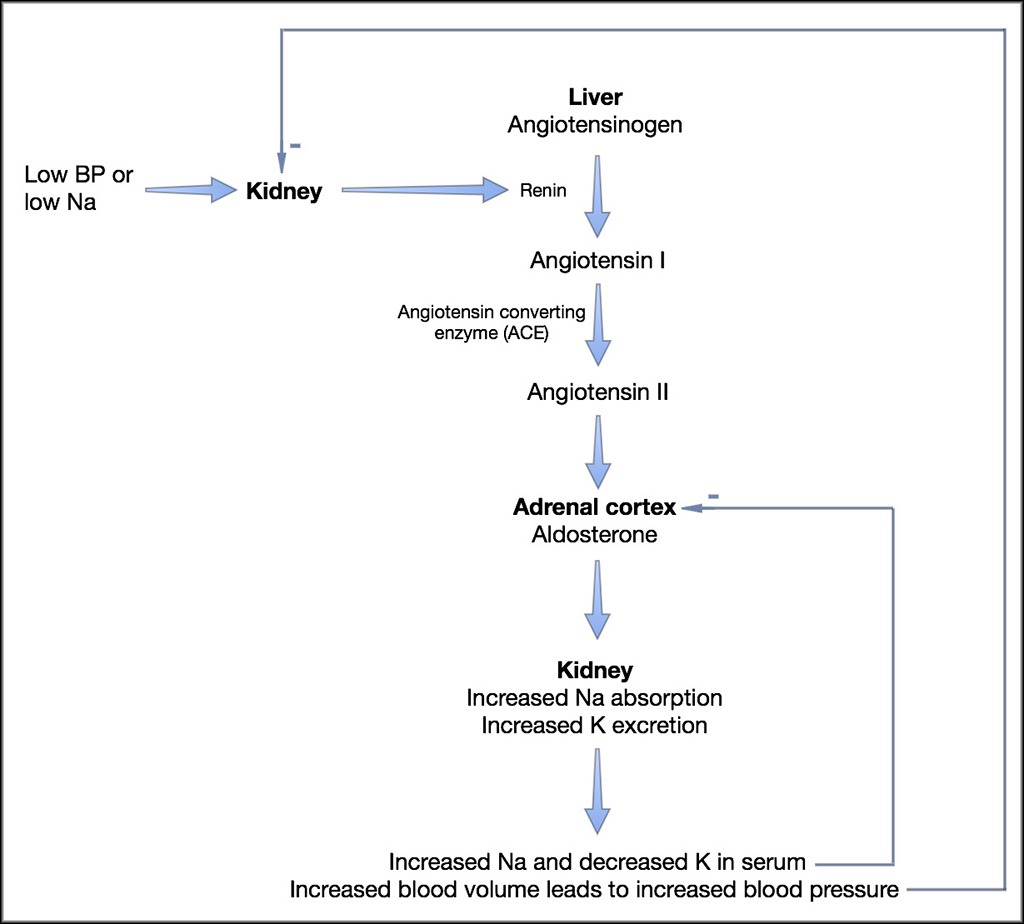
Fig. 1: Regulation of aldosterone secretion
The cortex of the adrenal gland has three zones and of these zones, two of them are important in the function of the adrenal gland. The outer zone – zona glomerulosa – secretes aldosterone and the next zone – zona fasciculata – secretes glucocorticoids. The most inner zone – zona reticularis – secretes sex hormones and they do not contribute to signs associated with hypoadrenocorticism. Aldosterone promotes sodium, chloride and water resorption, and potassium excretion in epithelial tissues. The primary site of effect is the renal tubule (proximal convoluted), however other tissues where an effect is exerted include the intestinal mucosa, salivary glands and sweat glands. Secretion of aldosterone itself is controlled by the renin-angiotensin system where angiotensin II is ultimately produced and acts directly on the adrenal gland to produce aldosterone (Fig. 1). Other stimulants of aldosterone include potassium through a transmembrane effect as well as, to a much lesser extent, ACTH hormone. Glucocorticoids are produced as a result of increased stress and ACTH secreted by the pituitary gland in response to corticotropin-releasing hormone (CRH), as well as cortisol metabolism. Cortisol affects almost every tissue in the body. Some of its actions include the effects on the vascular system, such as maintaining vascular tone; effects on metabolism such as stimulating gluconeogenesis and glycogenolysis; control over hormone release including ACTH, CRH, and vasopressin; control of the immune system by suppressing inflammatory response; stimulating erythrocytosis.
The cause of primary hypoadrenocorticism is most commonly thought to be immune-mediated destruction of the glands. Other causes include granulomatous disease seen with histoplasmosis or blastomycosis, hemorrhagic infarctions which can be seen with rodenticide toxicity or trauma, metastasis such as lymphoma, as well as amyloidosis. Iatrogenic causes can include surgery, treatment with Trilostane or Lysodren as well as rapid withdrawal of corticosteroids. Secondary hypoadrenocorticism usually is seen along with other neurologic and ophthalmologic signs and is caused by a primary deficiency of ACTH, as a result of lack of pituitary production or secondary deficiency of CRH production from the hypothalamus. Neoplasia, trauma and inflammation are common causes.
Signalment and clinical signs
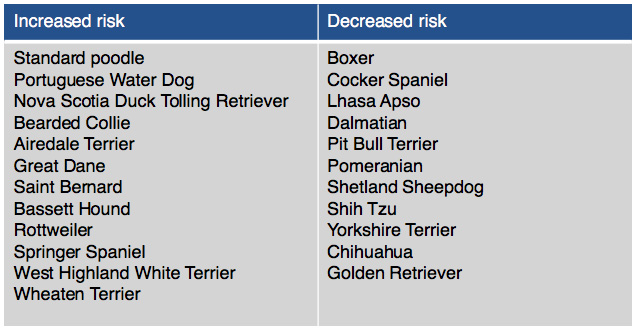
Table 1: Breeds reported to be either at increased or decreased risk of hypoadrenocorticism.
Dogs diagnosed with this disease are usually young to middle-aged and 69% are female. Mixed breed dogs are most commonly diagnosed, however there are breeds at higher risk for developing Addison’s disease. (Table 1).
There are no pathognomonic signs with hypoadrenocorticism and this disease is known as the great mimicker. It mimics signs seen with kidney, gastrointestinal (GI) and some infectious diseases. The signs can be waxing and waning or episodic but this occurs only in about 25% of dogs. Signs can include anorexia, lethargy, weight loss, weakness, vomiting or diarrhea, but can be as severe as collapse. Physical exam findings can also vary but can include weakness, thin body condition score, dehydration, shock, hypothermia, bradycardia, hypotension, and evidence of melena or hematochezia on rectal exam.
Baseline diagnostic testing
In patients with hypoadrenocorticism, a complete blood count may show normocytic, normochromic anemia (Hct 20-35%) secondary to bone marrow suppression from chronic hypocortisolism and this may not become apparent until the patient is rehydrated. Anemia may be significant especially in those patients with GI hemorrhage. Classically the complete blood count will lack a stress leukogram with a lymphocytosis and eosinophilia predominating. Ahbout 80% of patients with hypoadrenocorticism will have normal leukocyte counts. This finding alone should raise the suspicion for hypoadrenocorticism.
On chemistry profile, hyponatremia and hyperkalemia are classically found due to the lack of aldosterone production. A ratio below 27:1 should start to raise suspicion for hypoadrenocorticism, it has been found that a ratio of 24:1 is highly specific for the diagnosis. Other diseases can cause these electrolyte abnormalities (Tables 2 & 3). It is important to remember that these electrolyte changes are not always present in patients with hypoadrenocorticism. In atypical hypoadrenocorticism electrolytes will be normal. Other possible causes for hypoadrenocorticism with normal electrolytes include pituitary ACTH deficiency, lysodren or trilostane induced hypocortisolemia, or concurrent illness. Up to 30% of animals diagnosed with hypoadrenocorticism can have normal electrolytes. Increased BUN can be found especially in cases of dehydration, intestinal bleeding as well as an increased creatinine due to reduced renal perfusion. Urine specific gravity may be reduced due to impaired ability to concentrate urine due to chronic urinary sodium losses, medullary washout, impaired capacity for water resorption. Evidence of hepatic dysfunction may be present via hypoalbuminemia, hypoglycemia and hypocholesterolemia. Liver function testing may be slightly abnormal. Hypercalcemia and mild to moderate metabolic acidosis are also possible findings, especially the latter due to reduced tissue perfusion and impaired hydrogen excretion.
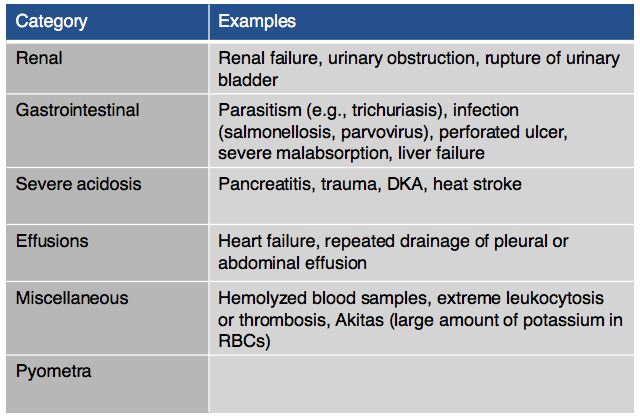 Table 2: Other causes of hyperkalemia |
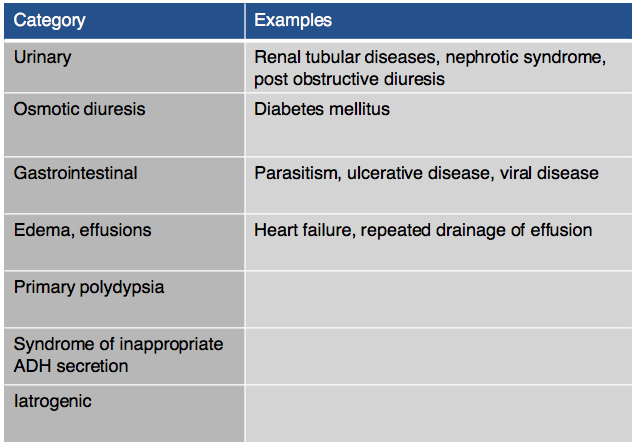 Table 3: Other causes of hyponatremia |
In animals with hypoadrenocorticism, thoracic radiographs may reveal megaesophagus as well as a flattened aorta and narrow posterior vena cava. The mechanism of megaesophagus is unclear. It is thought that this may be related to the effect of abnormal serum sodium and potassium concentrations on neuromuscular functions, or the lack of cortisol having an effect on muscle function.
Abdominal ultrasound findings include small adrenal gland size, although normal sized adrenal glands do not exclude hypoadrenocorticism as a diagnosis. In addition to this, evidence of more rare causes of hypoadrenocorticism may be found such as metastasis or granulomatous disease.
In some cases of hyperkalemia an EKG is indicated and possible findings include a slowed heart rate, peaked T–waves, widened QRS complex, and as potassium increases, one can diagnose an increased P-R interval, lack of p-wave, progressing to ventricular asystole or ventricular fibrillation.
Endocrine testing
Baseline cortisol concentrations can be used in some patients where hypoadrenocorticism is a suspicion. A cortisol concentration greater than 2 µg/dL is known with a 100% sensitivity to exclude the diagnosis of hypoadrenocorticism. However if the concentration is below 2 µg/dL, an ACTH stimulation test is needed to confirm the diagnosis.
The gold standard, and most commonly used method to diagnose hypoadrenocorticism, is an ACTH stimulation test where both pre and post values should be less than 2 µg/dL. This test does not distinguish primary versus secondary hypoadrenocorticism. To differentiate between these two an endogenous ACTH concentration can be submitted (careful handling instructions necessary) where a low concentration is diagnostic of secondary hypoadrenocorticism. To perform the ACTH stimulation test synthetic ACTH is administered. A dose of 5 µg/kg given intravenously or intramuscularly is a sufficient dose (max dose 250 µg/dog) with a post sample taken one hour after ACTH administration. It has recently been documented that there is no difference between the use of the lyophilized ACTH product intramuscularly and the liquid ACTH product given intravenously. If steroids need to be given prior to performing the ACTH stimulation test, it is important that Dexamethasone is used. Dexamethasone is not detected by cortisol assays and it has minimal effect on the post-ACTH cortisol concentration.
Treatment
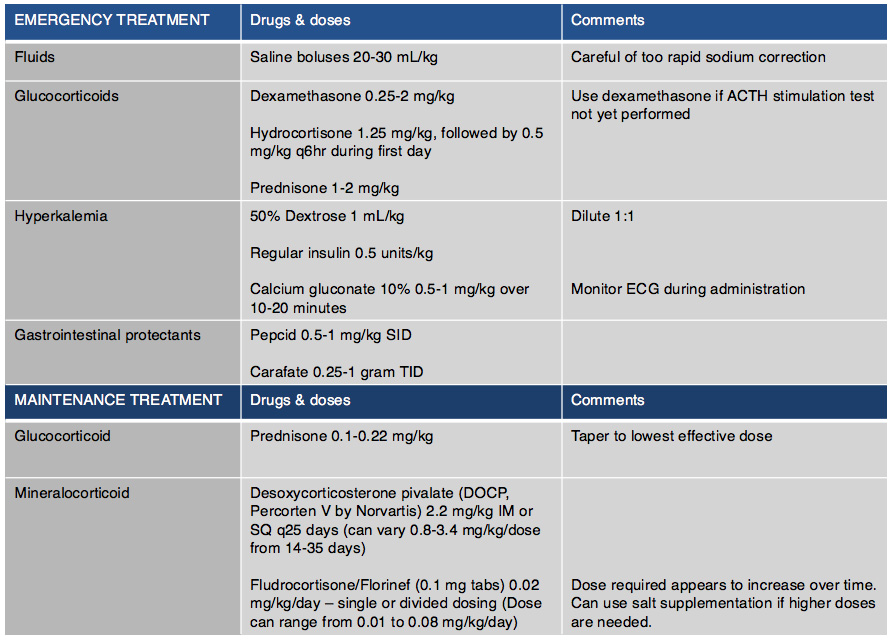
Table 4: Emergency and maintenance drug doses for treatment
Emergency treatment is often indicated in animals due to severe dehydration, hypotension and electrolyte abnormalities (Table 4). Saline boluses should be used although care should be given not to rapidly correct sodium blood concentrations as cerebral myelinosis may result. Glucocorticoids should be administered and dextrose should be given to treat hypoglycemia and help counteract hyperkalemia. If hyperkalemia is significant and life-threatening, one should also consider treating with insulin to lower potassium and calcium gluconate to protect the myocardium. DOCP may be given if there is strong clinical suspicion however ideally results of the ACTH stimulation test should be available.
Maintenance therapy will be tailored to each individual patient. Prednisone should be administered with every patient having the dose tapered to lowest dose that will control signs and avoid side effects. In some patients every other day prednisone dosing is adequate. It is important to increase dosage during times of stress by doubling the dose of steroid that the patient is getting for about one week after stress and then taper again over 2-4 weeks. In those patients where electrolyte abnormalities are diagnosed mineralocorticoid hormone supplementation is required. Desoxycorticosterone Pivalate (DOCP, Percorten V) is the most common drug used to supplement mineralocorticoids. Initially, electrolytes should be reevaluated one week after discharge, then at 14 and 25 days to assess electrolyte control and DOCP dose as well as interval needed between injections. At 14 days electrolytes should be normal and if they are not, the dose of DOCP should be increased by 5-10%. At day 25 electrolytes should be evaluated and the interval changed based upon control of electrolytes. When the interval appears to be greater than 30 days administration, the dose can be decreased by 10% at each dosing but careful electrolyte monitoring is necessary. After the first 2-3 months if an appropriate dose and interval is achieved owners can be shown how to administer the injection, and re-evaluation every 3-6 months should be scheduled. Fludrocortisone acetate (Florinef) can be used in place of DOCP if daily oral therapy is preferred. In some cases, when Florinef is used, prednisone therapy can be discontinued completely as it may have enough glucocorticoid therapy. However, some patients also may have severe glucocorticoid side effects and in those cases a switch to DOCP may be necessary. Electrolytes for patients using Florinef should be monitored weekly to every two weeks until electrolytes are stable, although continued monitoring 2-3 times per year is needed as increased dosing in first 6-18 months of treatment is often necessary. If it is determined that a switch to DOCP from Florinef is needed, then it is recommend to taper Florinef over 4-5 days beginning on the day that DOCP is given. Hydrocortisone is not good option for long term management of electrolytes as a high dose would be needed to get enough mineralocorticoid effect, and likely significant glucocorticoid side effects would be noted.
In those patients where electrolyte abnormalities are not noted, treatment with only prednisone is necessary. It is recommended however that electrolytes be monitored every 1-3 months at least for the first year post diagnosis.Owners should monitor for any signs of illness as certain patients do develop electrolyte abnormalities after initially being normal.
In general the prognosis for patients diagnosed with hypoadrenocorticism is excellent with a reported median survival time of 4.7 years. If poor response to therapy is noted electrolyte control should be evaluated as well for other underlying concurrent illnesses.
